PEDIATRIC SPINAL CORD TUMORS / SPINAL CORD TUMORS IN CHILDREN
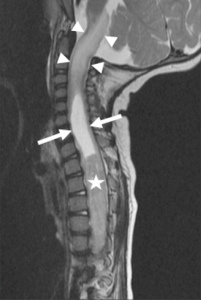
Spinal cord neoplasms are rare in the pediatric and adult patient population; however, spinal neoplasms are less common in children compared with adults. Only 0.5–1% of the central nervous system (CNS) neoplasms are located within the spinal cord. The vast majority (90%) of spinal cord neoplasms are glial tumors, and most of them are malignant. The rarity of these lesions is also demonstrated by the fact that of all CNS glial tumors, only 2–4% are located in the spinal cord. Finally, 35% of all intraspinal tumors are intrinsic spinal cord neoplasms.
All age groups may be affected, but spinal cord neoplasms are more frequent towards the end of the first decade and the beginning of the second decade of life. There is no gender predilection.
Diagnosis and clinical presentation
Unfortunately, most spinal cord tumors in children are diagnosed with a significant delay. The clinical symptoms may be misleading and vague. Many children present with a history of non-specific findings which progress slowly over time. In addition, children frequently have a long history of exacerbation and remission of symptoms, which is believed to result from fluctuant degrees of peritumoral spinal cord edema. So-called key clinical/neurological features include (a) progressive motor weakness, (b) progressive scoliosis, (c) gait disturbance, and (d) muscle rigidity with paraspinal muscle spasm. Sensory deficits are less common.
Back pain may be a leading clinical complaint; 25–30% of children with a spinal cord tumor suffer from recurrent episodes of back pain. Back pain can be classified as spinal pain (70%), root pain and tract pain. Spinal pain is characterized as a dull and aching pain, localized to the bone segments adjacent to the tumor. Spinal pain is also believed to result from focal distension of the dural sack by the tumor. Root pain and tract pain are less frequent. Root pain may mimic pain resulting from disk herniations. Tract pain is typically described as a vague, burning pain with ill-defined paresthesias. Tract pain likely results from a direct infiltration and compression of the spino-thalamic tracts within the spinal cord.
Children with back pain should be taken seriously, especially because back pain in healthy children without a history of recent trauma is very rare. Frequently, back pain in children is initially interpreted as so-called growth pain. Many of these children are in a phase of rapid growth and are physically highly active. Because most symptoms are non-specific and symptoms may wax and wane, diagnosis may be delayed. Physicians frequently do an array of diagnostic tests to rule out common causes for back pain, such as renal ultrasound for kidney stones, chest radiography for pneumonia and even spinal radiographic survey or bone scintigraphy for spondylodiscitis. When tests are negative, physicians may feel comfortable starting conservative treatments, which may result in a significant delay in diagnosis.
In rare cases, a spinal cord tumor can be missed because a limited magnetic resonance imaging (MRI) examination of the spine was requested, e.g. a lumbar spine MRI. Clinical symptoms may suggest a lumbar pathology. However, due to the complex segmentational development of the spinal cord and adjacent musculoskeletal derivates, a tumor may be located significantly higher than the symptoms suggest. Consequently, if a spinal MRI is performed the entire spinal cord should be imaged.
A smaller percentage of children with spinal cord neoplasms may present with symptoms of increased intracranial pressure. Several explanations are discussed: (a) cerebrospinal fluid (CSF) resorption may be impaired by a tumor-related elevation of the CSF proteins; (b) blockage of the foramen of magnum by a high cervical cord tumor; (c) by an intraspinal, subarachnoidal tumor hemorrhage; and (d) by subarachnoidal tumor seeding. Consequently, if an unexplained, new hydrocephalus is diagnosed, MRI of the spine to rule out spinal cord tumors should be considered.
Finally, diagnosis of spinal cord tumors may be delayed because these tumors are so rare that many physicians just do not think about it. Raising the awareness of this potentially devastating tumor is mandatory. Back pain, mild motor weakness or a mild gait disorder may be signs of a spinal cord tumor. The goal should be: diagnose aggressively (MRI) and treat as gently as possible as early as possible.
Neuroimaging
Several imaging modalities are available to diagnose spinal cord lesions. The high spatial resolution, the different image contrasts that can be generated, the combination of anatomical and functional data that can be collected and the lack of ionizing radiation make MRI the primary imaging modality for examining spinal cord lesions. Plain film radiography, myelography and ultrasonography are of little value in children. Computed tomography (CT) is contraindicated because of the high radiation dose if the entire spine is examined. Nuclear medicine studies are usually considered if an infectious process such as spondylodiscitis or an abscess is suspected.
MRI should always include the entire spine and if a focal spinal cord neoplasm is identified, the brain should be included to exclude additional or metastatic lesions. Spinal lesions may be metastatic lesions from an intracranial neoplasm. MRI protocols should include multiple planes taking advantage of different imaging contrasts like T1- and T2-weighted images. Contrast-enhanced images will increase diagnostic sensitivity. Finally, functional sequences including diffusion tensor imaging (DTI) and 1H magnetic resonance spectroscopy (MRS) may be helpful.
Postoperative and follow-up imaging may be challenging. Frequently, spinal cord neoplasms are treated by a combination of chemotherapy, radiotherapy and neurosurgical resection. For resection, extensive laminectomies may be necessary which require postoperative stabilization using hardware. This hardware, even when it is of non-ferromagnetic material, may significantly limit the diagnostic sensitivity of MRI. Imaging sequences should be carefully chosen to limit artifacts that can obscure residual or recurrent tumor. New MRI sequences that are less sensitive to susceptibility artifacts are progressively becoming available.
Imaging features
Imaging features suggestive of a spinal cord neoplasm include (a) focal signal alteration with cord expansion, (b) corresponding contrast enhancement and (c) tumoral and non-tumoral cysts. A focal T1- or T2-weighted signal alteration in combination with a cord expansion is the leading criterion for a spinal neoplasm. If a significant focal mass effect is lacking other causes for the signal alteration should be considered, such as transverse myelitis, demyelinating processes like multiple sclerosis and acute disseminated encephalomyelitis, sarcoidosis, arteriovenous fistula with intramedullary edema, or focal ischemia. The clinical presentation and evolution of symptoms may be helpful for differentiation. Cysts are frequently seen in spinal tumors. These cysts may be non-tumoral or tumoral. Non-tumoral cysts are frequently seen at the poles of the tumor; they may be a reactive dilatation of the central canal or represent fluid produced by the tumor itself. Characteristically, these non-tumoral cysts do not enhance. Tumoral cysts are typically within the tumor and show a peripheral enhancement. Tumoral cysts have to be resected because the cyst wall is likely invaded by tumor cells.
MRI usually allows differentiating neoplastic from non-neoplastic spinal cord lesions; however MRI is limited in defining tumor histology. Biopsy is frequently necessary.
Neoplasms of the spinal cord
Compared to the adult patient population, the same kind of spinal neoplasms are seen in children. However, the incidence and presentation differs. The vast majority of pediatric spinal cord tumors are glial tumors (90–95%). The most frequent are the pilocytic and anaplastic astrocytomas (60%) and ependymomas (30%). In children a benign subgroup of the ependymomas may be encountered: myxopapillary ependymoma. Non-glial tumors are much less frequent and include a variety of histologic subtypes such as hemangioblastoma, subependymoma, ganglioglioma, paraganglioma, lymphoma, primitive neuroectodermal tumor (PNET) neurocytoma, oligodendroglioma and metastases. As rule of thumb, it can be said that in children, astrocytomas are more frequent than ependymomas; in adults ependymomas are more frequent than astrocytomas. Some additional rules mention that ependymomas are more frequently seen in neurofibromatosis (NF) II patients while astrocytomas are more frequently seen in NF I patients. Finally, the higher the tumor location (cervical versus thoracic or lumbar), the more likely a syrinx will develop.
Spinal cord astrocytoma
Spinal astrocytomas are the most common spinal cord tumors in children. Their primary location is more rostral than in adults; 50% are in the cervico-thoracic region and usually affect a small number of segments. Astrocytomas most frequently present with pain and motor dysfunction, followed by gait disturbance, torticollis and scoliosis. Bowel and bladder dysfunction is uncommon due to their predominant cervical location. The majority are of low grade, WHO grade I and II (75–80%) including pilocytic and fibrillary type astrocytomas. Anaplastic astrocytoma (WHO III) and glioblastoma multiforme (WHO IV) are much rarer (20–25%, 0.2–1.5%).
Spinal astrocytomas rarely present with a clear cleavage plane between tumor and normal spinal cord because of the infiltrative characteristics; tumor cells extend along the scaffold of normal astrocytes. Tumor cysts, both polar and intratumoral, are seen in 20–40% of children, frequently accompanied by a rostral and/or caudal syrinx. Spinal cord astrocytomas are rarely hemorrhagic. On imaging the lesions are frequently eccentric within the spinal cord and may show an asymmetric spinal cord expansion. Lesions are typically T2-hyperintense, T1-iso- or hypointense and may show a mild to moderate contrast enhancement. Rarely, exophytic tumor components may be observed. On follow-up imaging, low grade spinal cord astrocytomas are typically stable or may show a slow progression. On DTI, long tract fibers may be interrupted. Tumor cysts frequently reveal a peripheral contrast enhancement. Careful analysis of the adjacent soft tissues as well as the brain is essential to rule out additional lesions that may suggest NF1.
Pilomyxoid astrocytomas represent a recently identified and described subtype of spinal cord astrocytomas. Imaging features are very similar to the classical astrocytomas, however they frequently present early with diffuse subarachnoidal dissemination.
Spinal cord ependymoma
Spinal ependymomas are considered to be the second most frequent spinal cord tumor. Several reports see ependymomas in strong competition with gangliogliomas for second place. Gangliogliomas resemble the signal characteristics of astrocytomas on imaging and are also frequently eccentric in location. Final diagnosis is usually made after biopsy.
Ependymomas are typically in a central location, around the central canal. This also explains the more frequently observed sensory symptoms due to their proximity to the spino-thalamic tracts. In addition, ependymomas may also present with back pain and motor weakness. Ependymomas are most frequently seen in the cervical spinal cord. Classical spinal ependymomas are usually low grade (WHO I, II) and are consequently slow growing. Ependymomas are less infiltrative than astrocytomas, they tend to compress and displace the adjacent spinal cord tissue. On DTI, the long tracts are typically displaced rather than interrupted. Frequently a clear cleavage plane may be seen on MRI. Polar cysts are common. In contrast to astrocytomas, tumor cysts are infrequent. Many ependymomas reveal a high vascularity with multiple small feedings vessels and a strong contrast enhancement. The high vascularity also increases the chance for intratumoral and subarachnoidal hemorrhage. A rim of T2-hypointense hemosiderin may be observed along the pole of the tumor, also known as ‘cap sign’.
Myxopapillary ependymoma is a special variant that differs by age of presentation and location from the classic ependymomas. Myxopapillary ependymomas encompass about 13% of all spinal ependymomas and are more common in male children. They typically arise from the ependymal glia of the filum terminal and are consequently seen predominantly in the area of the conus medullare and filum terminale. These tumors are frequently polylobulated, fill the spinal canal, may scallop the adjacent vertebral bodies and can produce mucin. Clinically, due to their low location, they present with lower back pain, leg weakness and sphincter dysfunction. Prognosis after complete resection is favorable.
Various non-glial tumors
Multiple tumors may be found along and within the spinal cord. These tumors are much less frequent and are usually diagnosed after biopsy or if a matching clinical history suggest another histologic subtype than astrocytoma or ependymoma, e.g. lymphomas, PNETs or oligodendrogliomas. Hemangioblastomas and metastatic disease deserve a separate discussion.
Hemangioblastoma are very infrequent, however their imaging findings may be characteristic. In addition, hemangioblastomas should be ruled out in children with Von Hippel Lindau (VHL) syndrome. VHL syndrome is an inherited multi-system disorder characterized by abnormal growth of blood vessels with an increased risk for hemangioblastomas. These tumors are typically intramedullary with extension into the intra- and extradural space. They may be solitary or multiple. Hemangioblastomas are highly vascular tumors with dilated feeding arteries and draining pial veins. They are at risk for acute subarachnoid hemorrhages or intramedullary hemorrhages. These tumors show an avid contrast enhancement. An associated syrinx is common as well as intramedullary venous edema. Spinal angiography remains a highly sensitive imaging modality to exclude multiplicity.
Metastatic disease can be classified as intramedullary or extramedullary. Intramedullary metastases are rare and may result from hematogenous spread or from direct extension from the leptomeninges. Children with intramedullary metastatic disease usually have a poor prognosis with rapid progression of clinical symptoms. Extramedullary, intradural metastasis are much more frequent and usually result from CSF seeding from primary CNS neoplasms which are in close proximity or within the CSF compartments. Typically CSF seeding is seen in, e.g. medulloblastomas, ependymomas, high grade astrocytomas, germinomas, or choroid plexus tumors. The lumbosacral region is most frequently affected with a nodular and irregular, contrast-enhancing thickening of the thecal sac and nerve roots. The surface of the spinal cord may be coated, also known as ‘sugar coating’. Contrast-enhanced T1-weighted sequences are mandatory., To avoid misinterpretation imaging should be performed prior to surgery. Intradural blood products from surgery may mimic tumor seeding.
Finally, various other non-neoplastic lesions may mimic malignancy. An intradural epidermoid in spinal dysraphia may be confused with a spinal neoplasm. Intramedullary abscesses may simulate tumor. Finally, paraspinal neoplasms may extend intraspinal, e.g. paraspinal rhabdomyosarcomas or extraosseous Ewing sarcomas. Correlation with the clinical history and dedicated anatomical and functional MRI usually solves this issue.
Acknowledgements
Cancer Imaging. 2009; 9(Special issue A): S45–S48.
Published online 2009 Oct 2. doi: 10.1102/1470-7330.2009.9012
PMCID: PMC2797470
PMID: 19965293
Pediatric tumors of the spine
Thierry A.G.M. Huisman
References
1. Epstein F, Epstein N. Intramedullary tumors of the spinal cord. In: McLone D, editor. Pediatric neurosurgery: surgery of the developing nervous system. New York: Grune and Stratton; 1982. pp. 529–40. [Google Scholar]
2. Koeller KK, Rosenblum RS, Morrison AL. Neoplasms of the spinal cord and filum terminale: radiologic-pathologic correlation. Radiographics. 2000;20:1721–49. [PubMed] [Google Scholar]
3. Brotchi J, Dewitte O, Levivier M, et al. A survey of 65 tumors within the spinal cord: surgical results and the importance of preoperative magnetic resonance imaging. J Neurosurg. 1991;29:651–7. [PubMed] [Google Scholar]
4. Rossi A, Gandolfo C, Morana G, Tortori-Donati P. Tumors of the spine in children. Neuroimaging Clin N Am. 2007;17:17–35. [PubMed] [Google Scholar]
Commentary
By Dr Prem Pillay
Pediatric spinal cord tumors may present with a delay in diagnosis. Close attention should be given to back pain without an injury. This should be seen by a Specialist and an MRI of the spine should be done to look for tumors. Modern treatment technologies including Microsurgery, Ultrasonic and Lasers for tumor removal are available. Radiotherapy including Stereotactic Radiotherapy are also treatment options. Some tumors may need chemotherapy.
Dr Prem Pillay is a Singapore Neurosurgeon who specializes in the treatment of Spinal tumors including pediatric spine tumors and who has worked and trained at the Cleveland Clinic-USA and the Hospital for Sick Children-Toronto. He was previously the Head of Neurosurgery at the Singapore General Hospital and is Currently the Medical Director of the Singapore Brain Spine Nerves Center doing his procedures at Mt Elizabeth Hospital and Mt Elizabeth Hospital Novena.
